14.5 Ribosomes and Protein Synthesis
Elizabeth Dahlhoff
Learning Objectives
By the end of this section, you will be able to do the following:
- Explain the role of ribosomes in protein synthesis.
- Describe the different steps in protein synthesis.
Protein Synthesis
The synthesis of proteins consumes more of a cell’s energy than any other metabolic process. In turn, proteins account for more mass than any other component of living organisms (with the exception of water), and proteins perform many different functions in a cell. The process of translation, or protein synthesis, involves the decoding of an mRNA message into a polypeptide product. Amino acids are covalently strung together by interlinking peptide bonds in lengths ranging from approximately 50 to more than 1,000 amino acid residues. Each individual amino acid has an amino group and a carboxyl group (COOH). Polypeptides are formed when the amino group of one amino acid forms an amide (i.e., peptide) bond with the carboxyl group of another amino acid. This reaction is catalyzed by ribosomes and generates one water molecule (it is a dehydration reaction).
The Protein Synthetic Machinery
In addition to the mRNA template, many molecules and macromolecules contribute to the process of translation. Translation requires the input of an mRNA template, ribosomes, tRNAs, and various enzymatic factors.
Ribosomes
Even before an mRNA is translated, a cell must invest energy to build its ribosomes. In an E. coli cell, there are between 10,000 and 70,000 ribosomes present at any given time. A ribosome is a complex macromolecule composed of ribosomal RNAs (rRNAs), and many polypeptides. In eukaryotes, the nucleolus is completely specialized for the synthesis and assembly of rRNAs.
Ribosomes are found in the cytoplasm of prokaryotes. In eukaryotes, they are also found in the cytoplasm and are sometimes associated with the outer surface of the rough endoplasmic reticulum. Mitochondria and chloroplasts also have their own ribosomes, which look more similar to prokaryotic ribosomes (and have similar drug sensitivities) than the ribosomes just outside their outer membranes in the cytoplasm.
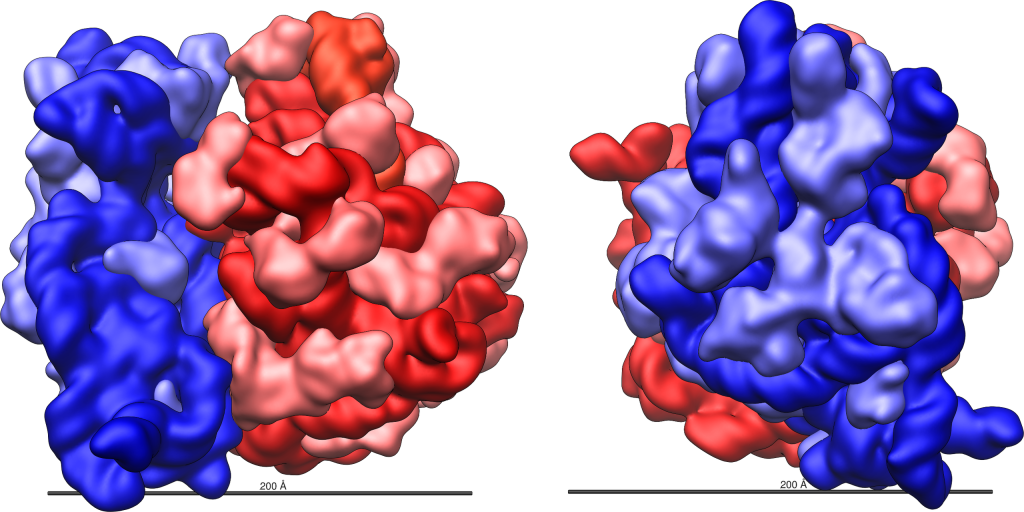
Ribosomes dissociate into large and small subunits when they are not synthesizing proteins and reassociate during the initiation of translation. In E. coli, the small subunit is described as 30S, and the large subunit is 50S, for a total of 70S (Svedberg units are not additive). Mammalian ribosomes have a small 40S subunit and a large 60S subunit, for a total of 80S. The small subunit is responsible for binding the mRNA template, whereas the large subunit sequentially binds tRNAs. Each mRNA molecule is simultaneously translated by many ribosomes (although in different locations along the mRNA), all synthesizing protein in the same direction: reading the mRNA from 5′ to 3′ and synthesizing the polypeptide from the N terminus to the C terminus.
tRNAs
The transfer RNAs (tRNAs) are structural RNA molecules that were transcribed from their corresponding genes by RNA polymerase III. Depending on the species, 40 to 60 types of tRNAs exist in the cytoplasm. Transfer RNAs serve as adaptor molecules. Each tRNA carries a specific amino acid and recognizes one or more of the mRNA codons that define the order of amino acids in a protein. Aminoacyl-tRNAs (tRNA with attached amino acid) bind to the ribosome and add the corresponding amino acid to the polypeptide chain. Therefore, tRNAs are the molecules that actually “translate” the language of RNA into the language of proteins.
Of the 64 possible mRNA codons—or triplet combinations of A, U, G, and C—three specify the termination of protein synthesis and 61 specify the addition of amino acids to the polypeptide chain. Of these 61, one codon (AUG) also encodes the initiation of translation. Each tRNA anticodon can base pair with one or more of the mRNA codons for its amino acid.
Aminoacyl tRNA Synthetases
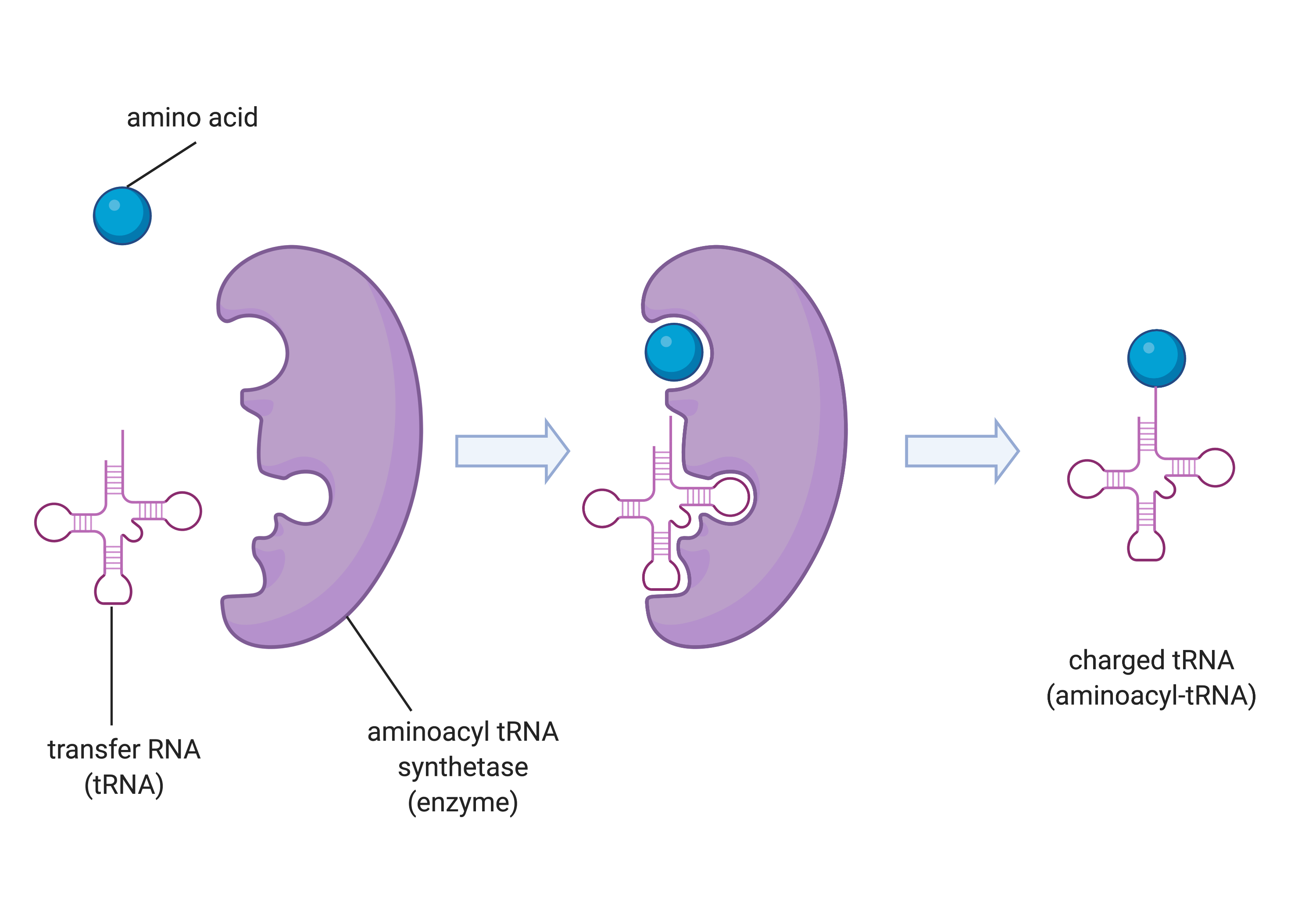
The process of pre-tRNA synthesis by RNA polymerase III only creates the RNA portion of the adaptor molecule. The corresponding amino acid must be added later, once the tRNA is processed and exported to the cytoplasm. Through the process of tRNA “charging,” each tRNA molecule is linked to its correct amino acid by one of a group of enzymes called aminoacyl tRNA synthetases. At least one type of aminoacyl tRNA synthetase exists for each of the 20 amino acids; the exact number of aminoacyl tRNA synthetases varies by species. The term “charging” is appropriate, since the high-energy bond that attaches an amino acid to its tRNA is later used to drive the formation of the peptide bond. Each tRNA is named for its amino acid.
https://youtu.be/morl5e-jBNk
Video 14.5.1. Ribosome by WEHImovies
Protein Synthesis
As with mRNA synthesis, protein synthesis can be divided into three phases: initiation, elongation, and termination. The process of translation is similar in prokaryotes and eukaryotes. Here we’ll explore how translation occurs in E. coli, a representative prokaryote, and specify any differences between prokaryotic and eukaryotic translation.
Initiation of Translation
Protein synthesis begins with the formation of an initiation complex. In E. coli, this complex involves the small 30S ribosome, the mRNA template, three initiation factors (IFs; IF-1, IF-2, and IF-3), and a special initiator tRNA, called tRNAMetf.
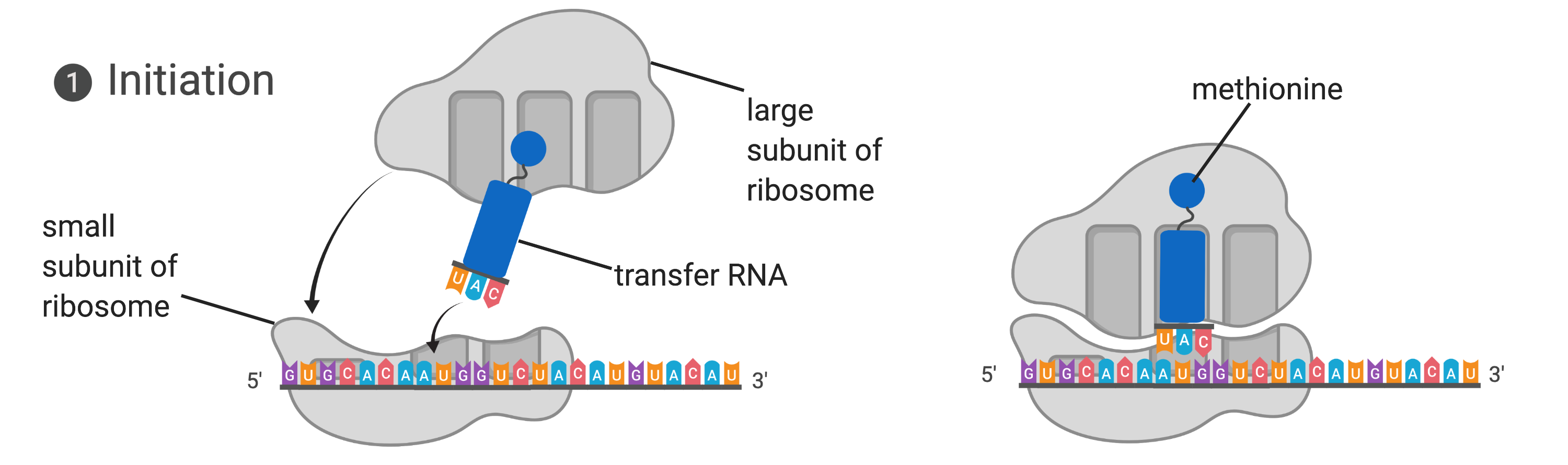
The small subunit of the ribosome is first to bind to the mRNA template at a specific sequence called the Shine-Dalgarno sequence. The initiator tRNA then interacts with the start codon AUG. This tRNA carries the amino acid methionine, which is formylated after its attachment to the tRNA. The formylation creates a “faux” peptide bond between the formyl carboxyl group and the amino group of the methionine. Binding of the fMet-tRNAMetf is mediated by the initiation factor IF-2. The fMet begins every polypeptide chain synthesized by E. coli, but it is usually removed after translation is complete. When an in-frame AUG is encountered during translation elongation, a non-formylated methionine is inserted by a regular Met-tRNAMet. After the formation of the initiation complex, the 30S ribosomal subunit is joined by the 50S subunit to form the translation complex. In eukaryotes, a similar initiation complex forms, comprising mRNA, the 40S small ribosomal subunit, eukaryotic IFs, and nucleoside triphosphates (GTP and ATP). The methionine on the charged initiator tRNA, called Met-tRNAi, is not formylated. However, Met-tRNAi is distinct from other Met-tRNAs in that it can bind IFs.
Once the appropriate AUG is identified, the other proteins dissociate, and the 60S subunit binds to the complex of Met-tRNAi, mRNA, and the 40S subunit. This step completes the initiation of translation in eukaryotes.
Practice Questions
Translation, Elongation, and Termination
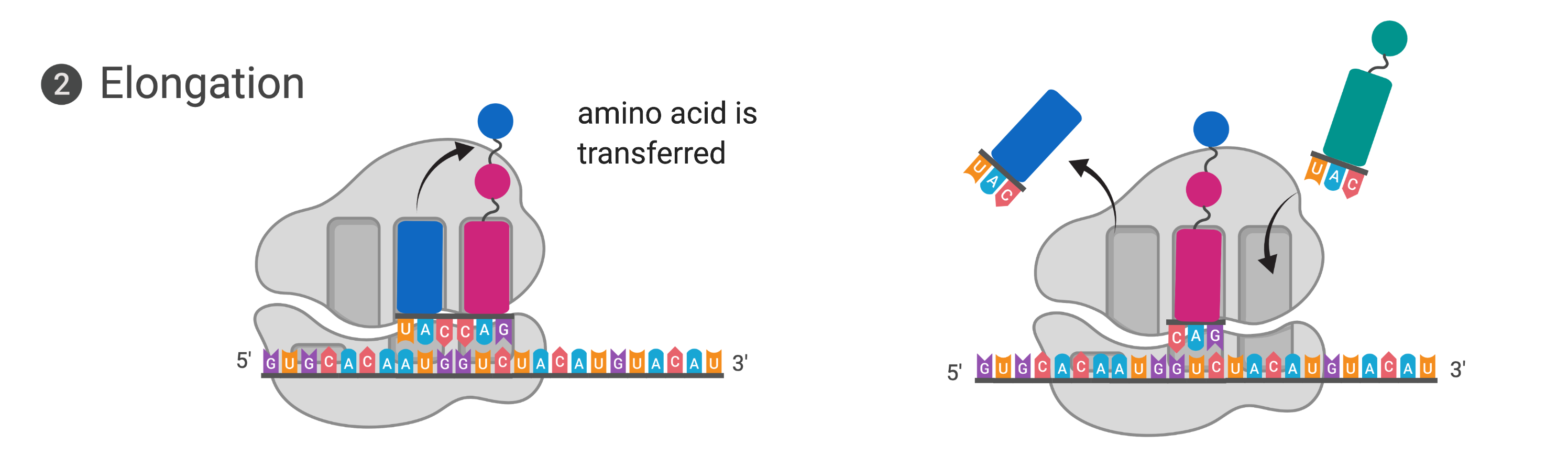
In prokaryotes and eukaryotes, the basics of elongation are the same, so we will review elongation from the perspective of E. coli. When the translation complex is formed, the tRNA binding region of the ribosome consists of three compartments. The A (aminoacyl) site binds incoming charged aminoacyl tRNAs. The P (peptidyl) site binds charged tRNAs carrying amino acids that have formed peptide bonds with the growing polypeptide chain but have not yet dissociated from their corresponding tRNA. The E (exit) site releases dissociated tRNAs so that they can be recharged with free amino acids. The initiating methionyl-tRNA, however, occupies the P site at the beginning of the elongation phase of translation in both prokaryotes and eukaryotes.
During translation elongation, the mRNA template provides tRNA binding specificity. As the ribosome moves along the mRNA, each mRNA codon comes into register, and specific binding with the anticodon of the corresponding charged tRNA anticodon is ensured. If mRNA were not present in the elongation complex, the ribosome would not bind to the correct tRNA, and translation would not be accurate.
Elongation proceeds with charged tRNAs sequentially entering and leaving the ribosome as each new amino acid is added to the polypeptide chain. Movement of a tRNA from A to P to E site is induced by conformational changes that advance the ribosome by three bases in the 3′ direction. The energy for each step along the ribosome is donated by elongation factors that hydrolyze GTP. GTP energy is required both for the binding of a new aminoacyl-tRNA to the A site and for its translocation to the P site after formation of the peptide bond. Peptide bonds form between the amino group of the amino acid attached to the A-site tRNA and the carboxyl group of the amino acid attached to the P-site tRNA. The formation of each peptide bond is catalyzed by peptidyl transferase, an RNA-based enzyme that is integrated into the 50S ribosomal subunit. The energy for each peptide bond formation is derived from the high-energy bond linking each amino acid to its tRNA. After peptide bond formation, the ribosome advances relative to the mRNA and tRNAs such that the A-site tRNA that now holds the growing peptide chain will be present in the P site, and the P-site tRNA that is now uncharged moves to the E site and is expelled from the ribosome. Amazingly, the E. coli translation apparatus takes only 0.05 seconds to add each amino acid, meaning that a 200-amino-acid protein can be translated in just 10 seconds.

Termination of translation occurs when a nonsense codon (UAA, UAG, or UGA) is encountered. Upon aligning with the A site, these nonsense codons are recognized by protein release factors that resemble tRNAs. The releasing factors in both prokaryotes and eukaryotes instruct peptidyl transferase to add a water molecule to the carboxyl end of the P-site amino acid. This reaction forces the P-site amino acid to detach from its tRNA, and the newly made protein is released. The small and large ribosomal subunits dissociate from the mRNA and from each other; they are recruited almost immediately into another translation initiation complex. After many ribosomes have completed translation, the mRNA is degraded so the nucleotides can be reused in another transcription reaction.
Video 14.5.2. Translation by Oxford Academic (Oxford University Press)
Protein Folding, Modification, and Targeting
During and after translation, individual amino acids may be chemically modified, signal sequences appended, and the new protein “folded” into a distinct three-dimensional structure as a result of intramolecular interactions. A signal sequence is a short sequence at the amino end of a protein that directs it to a specific cellular compartment. These sequences can be thought of as the protein’s “train ticket” to its ultimate destination, and are recognized by signal-recognition proteins that act as conductors. For instance, a specific signal sequence terminus will direct a protein to the mitochondria or chloroplasts (in plants). Once the protein reaches its cellular destination, the signal sequence is usually clipped off.
Many proteins fold spontaneously, but some proteins require helper molecules, called chaperones, to prevent them from aggregating during the complicated process of folding. Even if a protein is properly specified by its corresponding mRNA, it could take on a completely dysfunctional shape if abnormal temperature or pH conditions prevent it from folding correctly.
Practice Questions
Section Summary
The players in translation include the mRNA template, ribosomes, tRNAs, and various enzymatic factors. The small ribosomal subunit forms on the mRNA template either at the Shine-Dalgarno sequence (prokaryotes) or the 5′ cap (eukaryotes). Translation begins at the initiating AUG on the mRNA, specifying methionine. The formation of peptide bonds occurs between sequential amino acids specified by the mRNA template according to the genetic code. Charged tRNAs enter the ribosomal A site, and their amino acid bonds with the amino acid at the P site. The entire mRNA is translated in three-nucleotide “steps” of the ribosome. When a nonsense codon is encountered, a release factor binds and dissociates the components and frees the new protein. Folding of the protein occurs during and after translation.
Glossary
- aminoacyl tRNA synthetase
- enzyme that “charges” tRNA molecules by catalyzing a bond between the tRNA and a corresponding amino acid
- initiator tRNA
- in prokaryotes, called tRNAMetf; in eukaryotes, called tRNAi; a tRNA that interacts with a start codon, binds directly to the ribosome P site, and links to a special methionine to begin a polypeptide chain
- peptidyl transferase
- RNA-based enzyme that is integrated into the 50S ribosomal subunit and catalyzes the formation of peptide bonds
- polysome
- mRNA molecule simultaneously being translated by many ribosomes all going in the same direction
- signal sequence
- short tail of amino acids that directs a protein to a specific cellular compartment
- start codon
- AUG (or rarely, GUG) on an mRNA from which translation begins; always specifies methionine
Figure Descriptions
Figure 14.5.1. Two views of the E. coli 70S ribosome are shown side by side. In each view, the large 50S ribosomal subunit is depicted in shades of red, with the 23S rRNA in dark red, the 5S rRNA in orange-red, and ribosomal proteins in pink. The small 30S ribosomal subunit is shown in shades of blue, with the 16S rRNA in dark blue and ribosomal proteins in light blue. The left view shows the ribosome from one angle, and the right view shows it rotated to reveal a different orientation. Both include a horizontal scale bar indicating 200 Ångstroms (20 nm). [Return to Figure 14.5.1]
Figure 14.5.2. A three-step diagram depicts the charging of a tRNA molecule by aminoacyl tRNA synthetase. On the left, a blue sphere representing an amino acid and a pink cloverleaf-shaped transfer RNA (tRNA) are shown separately next to a purple, C-shaped enzyme labeled aminoacyl tRNA synthetase. In the middle panel, the amino acid and tRNA are both bound inside the enzyme. In the final panel on the right, the enzyme has released the tRNA now attached to the amino acid at its top, forming a charged tRNA (aminoacyl-tRNA). White arrows between panels indicate the progression of steps. [Return to Figure 14.5.2]
Figure 14.5.3. The diagram illustrates translation initiation in two panels. In the left panel, the small subunit of the ribosome is bound to a messenger RNA strand, represented as a sequence of colored letters, with an initiator tRNA carrying the anticodon UAC positioned above the start codon AUG. A large ribosomal subunit is shown above, ready to join. In the right panel, the large ribosomal subunit has joined the complex, enclosing the initiator tRNA within the ribosome. The tRNA carries the amino acid methionine, shown as a small blue sphere at the top of the tRNA. Arrows indicate the assembly process from the first to the second panel. [Return to Figure 14.5.3]
Figure 14.5.4. The diagram shows elongation in translation in two panels. In the left panel, the ribosome is bound to mRNA with three sites (E, P, and A) visible. A tRNA carrying the anticodon UAC and a blue amino acid is positioned in the A site next to a tRNA with a pink amino acid in the P site, and an arrow shows the blue amino acid being transferred to the growing polypeptide chain. In the right panel, the ribosome has shifted along the mRNA: the spent tRNA in the E site is exiting, the tRNA that was in the A site has moved to the P site carrying the growing chain, and the A site is empty and ready to accept the next tRNA. [Return to Figure 14.5.4]
Figure 14.5.5. The diagram shows termination of translation in two panels. In the left panel, the ribosome is bound to the mRNA, with a green release factor protein occupying the A site at a stop codon. The completed polypeptide chain, shown as a series of colored spheres, extends from the tRNA in the P site. In the right panel, the ribosome subunits have separated, the polypeptide chain has been released, and the mRNA is free, with the release factor and tRNA no longer bound to the ribosome. [Return to Figure 14.5.5]
Licenses and Attributions
This chapter, “Ribosomes and Protein Synthesis,” by Elizabeth Dahlhoff, is adapted from “The Protein Synthesis Machinery” and “The Mechanism of Protein Synthesis” in College Biology I by Melissa Hardy under a CC BY-NC 4.0 license and from “Ribosomes and Protein Synthesis” in Biology by Open Stax College (University of Hawaii) under a CC BY-NC 4.0 license. This work is licensed under a CC BY-NC 4.0 license.
Media Attributions
- Ribosome_shape © Vossman is licensed under a CC BY-SA (Attribution ShareAlike) license
- aminoacyl tRNA synthetase © Melissa Hardy is licensed under a CC BY-SA (Attribution ShareAlike) license
- Translation Initiation © Melissa Hardy is licensed under a CC BY (Attribution) license
- Translation Elongation © Melissa Hardy is licensed under a CC BY (Attribution) license
- Translation Termination © Melissa Hardy is licensed under a CC BY (Attribution) license
[latexpage]
Learning Objectives
By the end of this section, you will be able to:
- Compare homologous and analogous traits
- Discuss the purpose of cladistics
- Describe maximum parsimony
Scientists must collect accurate information that allows them to make evolutionary connections among organisms. Similar to detective work, scientists must use evidence to uncover the facts. In the case of phylogeny, evolutionary investigations focus on two types of evidence: morphologic (form and function) and genetic.
Two Options for Similarities
In general, organisms that share similar physical features and genomes tend to be more closely related than those that do not. Such features that overlap both morphologically (in form) and genetically are referred to as homologous structures; they stem from developmental similarities that are based on evolution. For example, the bones in the wings of bats and birds have homologous structures ([link]).
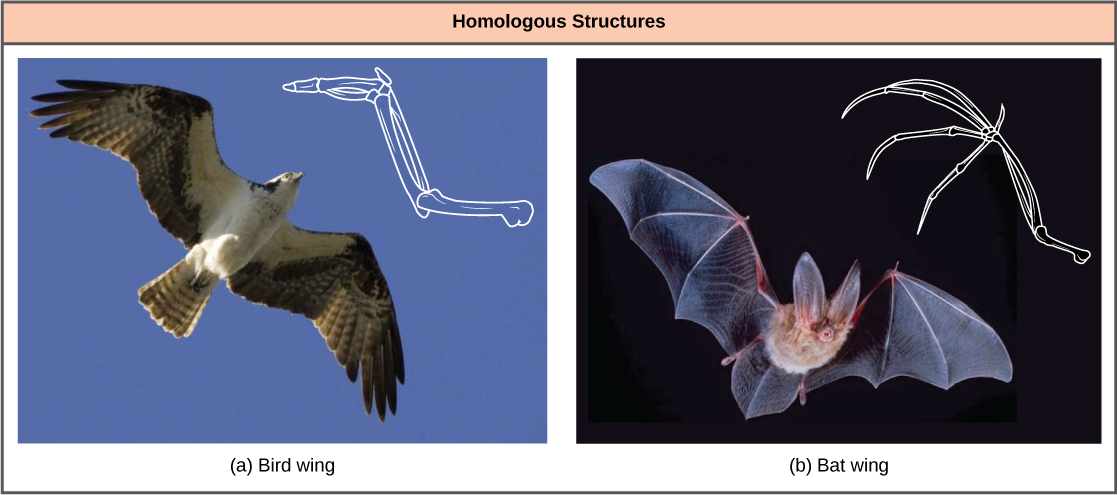
Notice it is not simply a single bone, but rather a grouping of several bones arranged in a similar way. The more complex the feature, the more likely any kind of overlap is due to a common evolutionary past. Imagine two people from different countries both inventing a car with all the same parts and in exactly the same arrangement without any previous or shared knowledge. That outcome would be highly improbable. However, if two people both invented a hammer, it would be reasonable to conclude that both could have the original idea without the help of the other. The same relationship between complexity and shared evolutionary history is true for homologous structures in organisms.
Misleading Appearances
Some organisms may be very closely related, even though a minor genetic change caused a major morphological difference to make them look quite different. Similarly, unrelated organisms may be distantly related, but appear very much alike. This usually happens because both organisms were in common adaptations that evolved within similar environmental conditions. When similar characteristics occur because of environmental constraints and not due to a close evolutionary relationship, it is called an analogy or homoplasy. For example, insects use wings to fly like bats and birds, but the wing structure and embryonic origin is completely different. These are called analogous structures ([link]).
Similar traits can be either homologous or analogous. Homologous structures share a similar embryonic origin; analogous organs have a similar function. For example, the bones in the front flipper of a whale are homologous to the bones in the human arm. These structures are not analogous. The wings of a butterfly and the wings of a bird are analogous but not homologous. Some structures are both analogous and homologous: the wings of a bird and the wings of a bat are both homologous and analogous. Scientists must determine which type of similarity a feature exhibits to decipher the phylogeny of the organisms being studied.


This website has several examples to show how appearances can be misleading in understanding the phylogenetic relationships of organisms.
Molecular Comparisons
With the advancement of DNA technology, the area of molecular systematics, which describes the use of information on the molecular level including DNA analysis, has blossomed. New computer programs not only confirm many earlier classified organisms, but also uncover previously made errors. As with physical characteristics, even the DNA sequence can be tricky to read in some cases. For some situations, two very closely related organisms can appear unrelated if a mutation occurred that caused a shift in the genetic code. An insertion or deletion mutation would move each nucleotide base over one place, causing two similar codes to appear unrelated.
Sometimes two segments of DNA code in distantly related organisms randomly share a high percentage of bases in the same locations, causing these organisms to appear closely related when they are not. For both of these situations, computer technologies have been developed to help identify the actual relationships, and, ultimately, the coupled use of both morphologic and molecular information is more effective in determining phylogeny.
Why Does Phylogeny Matter?Evolutionary biologists could list many reasons why understanding phylogeny is important to everyday life in human society. For botanists, phylogeny acts as a guide to discovering new plants that can be used to benefit people. Think of all the ways humans use plants—food, medicine, and clothing are a few examples. If a plant contains a compound that is effective in treating cancer, scientists might want to examine all of the relatives of that plant for other useful drugs.
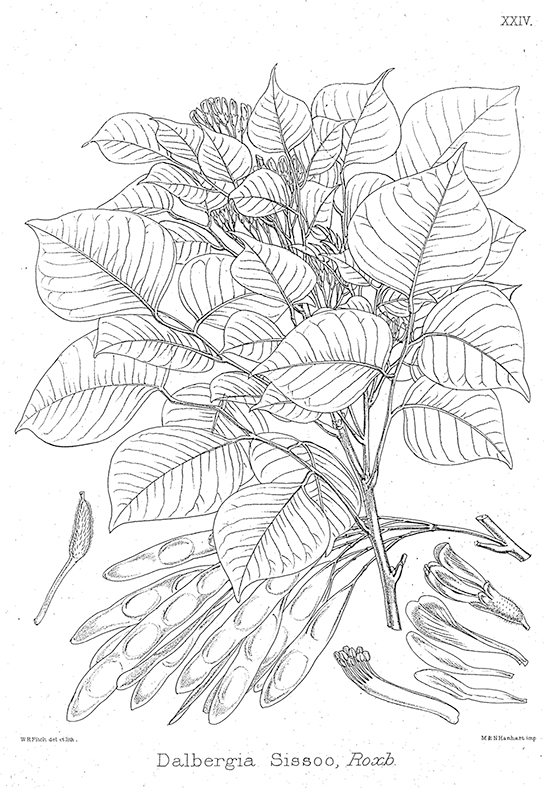
A research team in China identified a segment of DNA thought to be common to some medicinal plants in the family Fabaceae (the legume family) and worked to identify which species had this segment ([link]). After testing plant species in this family, the team found a DNA marker (a known location on a chromosome that enabled them to identify the species) present. Then, using the DNA to uncover phylogenetic relationships, the team could identify whether a newly discovered plant was in this family and assess its potential medicinal properties.
Building Phylogenetic Trees
How do scientists construct phylogenetic trees? After the homologous and analogous traits are sorted, scientists often organize the homologous traits using a system called cladistics. This system sorts organisms into clades: groups of organisms that descended from a single ancestor. For example, in [link], all of the organisms in the orange region evolved from a single ancestor that had amniotic eggs. Consequently, all of these organisms also have amniotic eggs and make a single clade, also called a monophyletic group. Clades must include all of the descendants from a branch point.
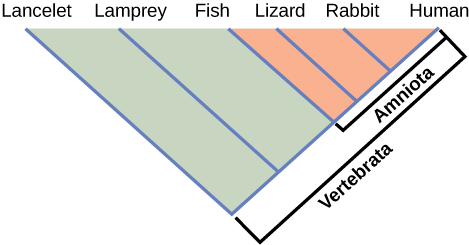
Which animals in this figure belong to a clade that includes animals with hair? Which evolved first, hair or the amniotic egg?
<!--<para> Rabbits and humans belong in the clade that includes animals with hair. The amniotic egg evolved before hair because the Amniota clade is larger than the clade that encompasses animals with hair.-->
Clades can vary in size depending on which branch point is being referenced. The important factor is that all of the organisms in the clade or monophyletic group stem from a single point on the tree. This can be remembered because monophyletic breaks down into “mono,” meaning one, and “phyletic,” meaning evolutionary relationship. [link] shows various examples of clades. Notice how each clade comes from a single point, whereas the non-clade groups show branches that do not share a single point.
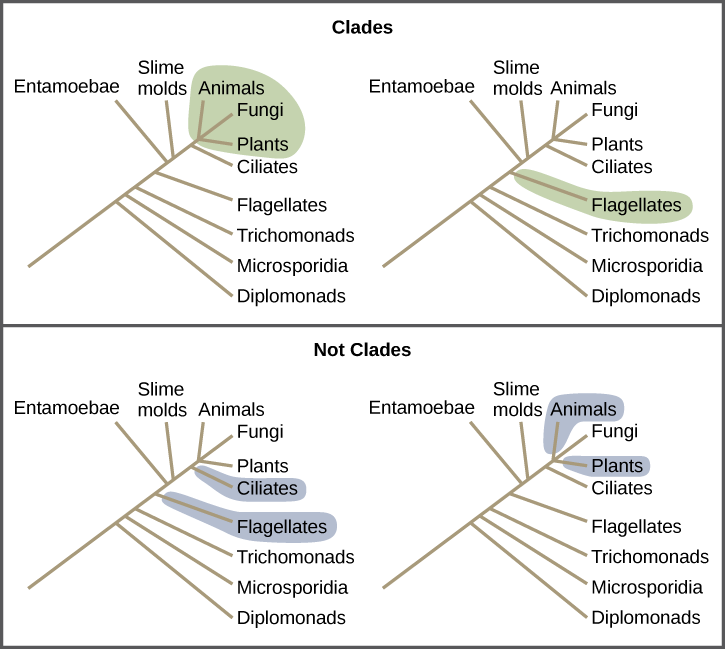
What is the largest clade in this diagram?
<!--<para> The largest clade encompasses the entire tree.-->
Shared Characteristics
Organisms evolve from common ancestors and then diversify. Scientists use the phrase “descent with modification” because even though related organisms have many of the same characteristics and genetic codes, changes occur. This pattern repeats over and over as one goes through the phylogenetic tree of life:
- A change in the genetic makeup of an organism leads to a new trait which becomes prevalent in the group.
- Many organisms descend from this point and have this trait.
- New variations continue to arise: some are adaptive and persist, leading to new traits.
- With new traits, a new branch point is determined (go back to step 1 and repeat).
If a characteristic is found in the ancestor of a group, it is considered a shared ancestral character because all of the organisms in the taxon or clade have that trait. The vertebrate in [link] is a shared ancestral character. Now consider the amniotic egg characteristic in the same figure. Only some of the organisms in [link] have this trait, and to those that do, it is called a shared derived character because this trait derived at some point but does not include all of the ancestors in the tree.
The tricky aspect to shared ancestral and shared derived characters is the fact that these terms are relative. The same trait can be considered one or the other depending on the particular diagram being used. Returning to [link], note that the amniotic egg is a shared ancestral character for the Amniota clade, while having hair is a shared derived character for some organisms in this group. These terms help scientists distinguish between clades in the building of phylogenetic trees.
Choosing the Right Relationships
Imagine being the person responsible for organizing all of the items in a department store properly—an overwhelming task. Organizing the evolutionary relationships of all life on Earth proves much more difficult: scientists must span enormous blocks of time and work with information from long-extinct organisms. Trying to decipher the proper connections, especially given the presence of homologies and analogies, makes the task of building an accurate tree of life extraordinarily difficult. Add to that the advancement of DNA technology, which now provides large quantities of genetic sequences to be used and analyzed. Taxonomy is a subjective discipline: many organisms have more than one connection to each other, so each taxonomist will decide the order of connections.
To aid in the tremendous task of describing phylogenies accurately, scientists often use a concept called maximum parsimony, which means that events occurred in the simplest, most obvious way. For example, if a group of people entered a forest preserve to go hiking, based on the principle of maximum parsimony, one could predict that most of the people would hike on established trails rather than forge new ones.
For scientists deciphering evolutionary pathways, the same idea is used: the pathway of evolution probably includes the fewest major events that coincide with the evidence at hand. Starting with all of the homologous traits in a group of organisms, scientists look for the most obvious and simple order of evolutionary events that led to the occurrence of those traits.

Head to this website to learn how maximum parsimony is used to create phylogenetic trees.
These tools and concepts are only a few of the strategies scientists use to tackle the task of revealing the evolutionary history of life on Earth. Recently, newer technologies have uncovered surprising discoveries with unexpected relationships, such as the fact that people seem to be more closely related to fungi than fungi are to plants. Sound unbelievable? As the information about DNA sequences grows, scientists will become closer to mapping the evolutionary history of all life on Earth.
Section Summary
To build phylogenetic trees, scientists must collect accurate information that allows them to make evolutionary connections between organisms. Using morphologic and molecular data, scientists work to identify homologous characteristics and genes. Similarities between organisms can stem either from shared evolutionary history (homologies) or from separate evolutionary paths (analogies). Newer technologies can be used to help distinguish homologies from analogies. After homologous information is identified, scientists use cladistics to organize these events as a means to determine an evolutionary timeline. Scientists apply the concept of maximum parsimony, which states that the order of events probably occurred in the most obvious and simple way with the least amount of steps. For evolutionary events, this would be the path with the least number of major divergences that correlate with the evidence.
Art Connections
[link] Which animals in this figure belong to a clade that includes animals with hair? Which evolved first, hair or the amniotic egg?
[link] Rabbits and humans belong in the clade that includes animals with hair. The amniotic egg evolved before hair because the Amniota clade is larger than the clade that encompasses animals with hair.
Review Questions
Which statement about analogies is correct?
- They occur only as errors.
- They are synonymous with homologous traits.
- They are derived by similar environmental constraints.
- They are a form of mutation.
C
What do scientists use to apply cladistics?
- homologous traits
- homoplasies
- analogous traits
- monophyletic groups
A
What is true about organisms that are a part of the same clade?
- They all share the same basic characteristics.
- They evolved from a shared ancestor.
- They usually fall into the same classification taxa.
- They have identical phylogenies.
B
Why do scientists apply the concept of maximum parsimony?
- to decipher accurate phylogenies
- to eliminate analogous traits
- to identify mutations in DNA codes
- to locate homoplasies
A
Free Response
Dolphins and fish have similar body shapes. Is this feature more likely a homologous or analogous trait?
Dolphins are mammals and fish are not, which means that their evolutionary paths (phylogenies) are quite separate. Dolphins probably adapted to have a similar body plan after returning to an aquatic lifestyle, and, therefore, this trait is probably analogous.
Why is it so important for scientists to distinguish between homologous and analogous characteristics before building phylogenetic trees?
Phylogenetic trees are based on evolutionary connections. If an analogous similarity were used on a tree, this would be erroneous and, furthermore, would cause the subsequent branches to be inaccurate.
Describe maximum parsimony.
Maximum parsimony hypothesizes that events occurred in the simplest, most obvious way, and the pathway of evolution probably includes the fewest major events that coincide with the evidence at hand.
Glossary
- analogy
- (also, homoplasy) characteristic that is similar between organisms by convergent evolution, not due to the same evolutionary path
- cladistics
- system used to organize homologous traits to describe phylogenies
- maximum parsimony
- applying the simplest, most obvious way with the least number of steps
- molecular systematics
- technique using molecular evidence to identify phylogenetic relationships
- monophyletic group
- (also, clade) organisms that share a single ancestor
- shared ancestral character
- describes a characteristic on a phylogenetic tree that is shared by all organisms on the tree
- shared derived character
- describes a characteristic on a phylogenetic tree that is shared only by a certain clade of organisms
RNA that carries activated amino acids to the site of protein synthesis on the ribosome
enzyme that “charges” tRNA molecules by catalyzing a bond between the tRNA and a corresponding amino acid

{kind=link}